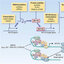

![ERIC2 PCR profiles of seven Spanish and three French isolates of ESBL-producing E. coli . Lane 1, molecular weight marker. Lanes 2 –8, Spanish isolates with strain FV7591 in lane 6. Lanes 9– 11, French isolates [strain HBS1 (lane 9), strain HDE2 (lane 10) and strain TE1 (lane 11)]. A uniform profile was found among these isolates except for the Spanish strain FV7591 (CTX-M-1).](https://www.researchgate.net/profile/Jorge-Blanco-9/publication/5771885/figure/fig1/AS:326192759623680@1454781822084/ERIC2-PCR-profiles-of-seven-Spanish-and-three-French-isolates-of-ESBL-producing-E-coli_Q320.jpg)



Content uploaded by Jorge Blanco
Author content
All content in this area was uploaded by Jorge Blanco on Feb 06, 2016
Content may be subject to copyright.
Intercontinental emergence of Escherichia coli clone O25:H4-ST131
producing CTX-M-15
Marie-He
´
le
`
ne Nicolas-Chanoine
1,2
*, Jorge Blanco
3
,Ve
´
ronique Leflon-Guibout
1
, Raphael Demarty
1
,
Maria Pilar Alonso
4
, Maria Manuela Canic¸a
5
, Yeon-Joon Park
6
, Jean-Philippe Lavigne
7
,
Johann Pitout
8
and James R. Johnson
9
1
Service de Microbiologie, Ho
ˆ
pital AP-HP Beaujon, 92110 Clichy, France;
2
Inserm, U-773, Faculte
´
de Me
´
decine
D. Diderot, Universite
´
Paris 7, Paris, France;
3
E. coli Reference Laboratory, Department of Microbiology and
Parasitology, Faculty of Veterinary Science, University of Santiago de Compostela, Lugo, Spain;
4
Laboratory of
Clinical Microbiology, Complejo Hospitalario Xeral-Calde, Lugo, Spain;
5
Antibiotic Resistance Unit, National
Institute of Health Dr Ricardo Jorge, Lisbon, Portugal;
6
Department of Clinical Pathology, College of Medicine,
The Catholic University of Korea, Kangnam St Mary’s Hospital, Seoul, South Korea;
7
Laboratoire de
Bacte
´
riologie, Virologie et Parasitologie, CHU de Nı
ˆ
mes, Nı
ˆ
mes, France;
8
Calgary Laboratory Services and
Department of Pathology and Laboratory Medicine, University of Calgary, Calgary, Alberta, Canada;
9
Veterans
Affairs Medical Center and University of Minnesota, Minneapolis, MN, USA
Received 29 August 2007; returned 12 October 2007; revised 19 October 2007; accepted 6 November 2007
Background: Concomitant with the recent emergence of CTX-M-type extended-spectrum b-lactamases
(ESBLs), Escherichia coli has become the enterobacterial species most affected by ESBLs. Multiple
locales are encountering CTX-M-positive E. coli, including specifically CTX-M-15. To gain insights into
the mechanism underlying this phenomenon, we assessed clonality and diversity of virulence profiles
within an international collection of CTX-M-15-positive E. coli.
Methods: Forty-one ESBL-positive E. coli isolates from eight countries and three continents (Europe,
Asia and North America) were selected for study based on suspected clonality. Phylogenetic group,
ERIC2 PCR profile, O H serotype, AmpC variant and antibiotic susceptibility were determined.
Multilocus sequence typing (MLST) and PFGE provided additional discrimination. Virulence potential
was inferred by detection of 46 virulence factor (VF) genes.
Results: Thirty-six (88%) of the 41 E. coli isolates exhibited the same set of core characteristics: phylo-
genetic group B2, ERIC2 PCR profile 1, serotype O25:H4, AmpC EC6, ciprofloxacin resistance and
MLST profile ST131. By PFGE, the 36 isolates constituted one large cluster at the 68% similarity level;
this comprised 17 PFGE groups (defined at 85% similarity), some of which included strains from differ-
ent countries. The 36 isolates exhibited highly (91% to 100%) similar VF profiles.
Conclusions: We describe a broadly disseminated, CTX-M-15-positive and virulent E. coli clonal group
with highly homogeneous virulence genotypes and subgroups exhibiting highly similar PFGE profiles,
suggesting recent emergence. Understanding how this clone has emerged and successfully dissemi-
nated within the hospital and community, including across national boundaries, should be a public
health priority.
Keywords: enterobacteria, E. coli , multidrug resistance
Introduction
Escherichia coli, a universal commensal of humans and several
animal species, is also one of the most common enterobacterial
species causing extraintestinal infections in these same hosts.
E. coli infections are becoming increasingly difficult to treat
because of emerging antimicrobial resistance, most recently to
expanded-spectrum cephalosporins, which is usually due to the
.....................................................................................................................................................................................................................................................................................................................................................................................................................................
*Corresponding author. Tel: þ33-1-40-87-56-06; Fax: þ33-1-40-87-05-50; E-mail: mhn.chanoine@bjn.aphp.fr
Journal of Antimicrobial Chemotherapy (2008) 61, 273–281
doi:10.1093/jac/dkm464
Advance Access publication 11 December 2007
.....................................................................................................................................................................................................................................................................................................................................................................................................................................
273
# The Author 2007. Published by Oxford University Press on behalf of the British Society for Antimicrobial Chemotherapy. All rights reserved.
For Permissions, please e-mail: journals.permissions@oxfordjournals.org
at Universidad de Santiago de Compostela on February 6, 2016http://jac.oxfordjournals.org/Downloaded from
production of extended-spectrum b-lactamases (ESBLs).
1
The
earliest ESBLs, which were first reported in 1985, consisted of
plasmid-mediated TEM-1, TEM-2 and SHV-1 derivatives and
were primarily a hospital-based problem.
1
However, since 2000,
ESBLs increasingly have also appeared in th e community.
2
This
phenomenon coincided with the emergence of a new group of
plasmid-mediated ESBLs, namely the CTX-M enzymes, which
seem to be taking over as the main ESBL type in some
locales.
3,4
Multiple locales are encountering CTX-M-positive E. coli
clinical isolates, including specifically CTX-M-15, which is one
of the more than 60 variants described in this enzyme group and
is able to efficiently hydrolyse not only cefotaxime but also cef-
tazidime.
3–8
The widespread occurrence of CTX-M-15-positive
E. coli could have two alternative explanations. That is, the cor-
responding plasmids or other mobile genetic elements surround-
ing the plasmid-mediated bla
CTX-M-15
gene may be moving from
strain to strain through the E. coli population.
3,9
Alternatively,
the strains themselves may be spreading in a clonal fashion, as
has been described for methicillin-resistant Staphylococcus
aureus, penicillin-resistant Streptococcus pneumoniae and
certain clonal groups of trimethoprim/sulfamethoxazole-resistant
E. coli (i.e. ‘clonal group A’ and E. coli O15:K52:H1).
10 – 13
While some articles have reported on the similarity of
CTX-M-15-encoding plasmids harboured by strains in different
locations, in the present study, we focused on strain genetic
background to assess the extent of clonality within a collection
of international CTX-M-positive E. coli isolates that we sus-
pected were clonally related to a group of French CTX-M-15
isolates of serogroup O25 previously studied in our labora-
tory.
6,14,15
We also sought to assess these strains’ molecularly
inferred virulence potential, a possible contributor (in addition
to antimicrobial resistance) to their recent emergence and disse-
mination as successful pathogens.
Materials and methods
Isolate collection
While we observed in France that CTX-M-15 was the most
common CTX-M enzyme and that the great majority of epide-
miologically unrelated, CTX-M-15-positive E. coli isolates dis-
played an identical genetic background, we also observed that
different studies performed in different countries found
CTX-M-15 as the most common CTX-M-type enzyme in E coli.
These concomitant observations pushed us to see whether this
worldwide outbreak of CTX-M-15-producing E. coli was due to
the spread of a clonal strain as found in France. Therefore, a
total of 41 recent human E. coli isolates that were known (n ¼
34) or presumed (n ¼ 7) to produce CTX-M-15, from th ree con-
tinents (Europe, Asia and North America) and eight countries
(France, Portugal, Spain, Switzerland, Lebanon, India, Korea
and Canada) were studied. They were selected because they
either were known to be clonal (the 13 French isolates) or, if
from outside France, were suspected of being related to the
French isolates based on CTX-M-15 production, the O25
antigen, the phylogenetic group B2 and/or ciprofloxacin resist-
ance. Previously published isolates included those from Canada
(n ¼ 6), India (n ¼ 2), Korea (n ¼ 2), Lebanon (n ¼ 4) and
Portugal (n ¼ 5).
16 – 20
As indicated in Table 1, they included
community-, hospital- and nursing-home-acquired isolates.
Except for the Lebanese and two French strains that were diges-
tive tract colonizers, the isolates were obtained from clinical
samples: primarily urine but also blood, sputum, intra-abdominal
pus and ascites (Table 1).
Relevant characteristics of the isolates that were known prior
to this study are italicized in Table 1. Notably, the French iso-
lates [including a strain (TE1) previously reported as responsible
for an outbreak in a long-term care facility] were previously
shown to produce CTX-M-15 and to exhibit the same genomic
PCR profile, O antigen (O25), chromosomal cephalosporinase
variant (AmpC EC6) and ciprofloxacin phenotype
(resistant).
15,21
b
-Lactamase determination
CTX-M-type b-lactamase genes were identified as previously
described.
21
Briefly, two sets of primers were used, allowing
amplification and sequencing of any type of bla
CTX-M
gene
(primer set 1) versus specific bla
CTX-M
variants (primer set 2).
To determine an isolate’s ampC variant, DNA amplification was
done using primers VL1A
(5
0
-TGCACGATCTGAAAATCCAC-3
0
) and VL2A
(5
0
-AGCAGGCGCATAAATGTTTC-3
0
) under standard PCR
conditions, with a T
m
of 428C, which yielded a fragment of
1398 bp. Direct sequencing of the PCR product was performe d
using these PCR primers and two additional primers, VL1S
(5
0
-TATCTTCA
ATGGTCG-3
0
) and VL2S (5
0
-TGCATGGGCTCCAGG-3
0
). The
ampC nucleotide sequences and deduced protein sequences were
analysed by using software available at the Biosupport web site
(http://bioinfo.hku.hk/). These were then compared with those
available in GenBank by using Blast sequence software (htpp://
www.ncbi.nlm.nih.gov). The new AmpC peptide sequences were
named EC66 and EC68 and their corresponding genes were
deposited in GenBank under accession numbers EF507686 and
EF507687, respectively.
ERIC2 PCR profiles
ERIC2 PCR profiles, which are strain-specific banding patterns
obtained by amplifying multiple anonymous regions of the
genome using repetitive element-based primers, were generated
as previously described, with bacterial lysates used as template
DNA.
22
Profiles were defined as different when they exhibited at
least one high intensity band difference according to visual
inspection.
Phylogenetic group
Determination of major E. coli phylogenetic group (A, B1, B2
and D) was done by multiplex PCR.
23
Serotyping
The determination of O and H antigens was carried out by using
the method previously described by Guine
´
e et al.,
24
in which all
available O (O1–O185) and H (H1– H56) antisera were tested.
All antisera were obtained and absorbe d with the corresponding
cross-reacting antigens to remove the non-specific agglutinins.
The O25 antigen was also determined by a PCR-based
method.
25
Nicolas-Chanoine et al.
274
at Universidad de Santiago de Compostela on February 6, 2016http://jac.oxfordjournals.org/Downloaded from
Table 1. Clinical, bacterial and molecular characteristics of the 41 studied isolates of ESBL-producing E. coli
Country/
isolate
Sample/
acquisition site
Epidemic
(Ep) or
sporadic
(Sp) CTX-M-type
ERIC2
PCR
profile
Phylogenetic
group
AmpC
type Serotype
Antimicrobial susceptibility
CIP GEN AMK TET CHL SXT
France
MECB5 urine/C Sp M15 1 B2 EC6 O25:H4 R RRRSR
Vlab2 urine/C Sp M15 1 B2 EC6 O25:H4 R RSRSR
VA1 catheter/C Sp M15 1 B2 EC6 O25:H4 R RRRSR
VB6 urine/C Sp M15 1 B2 EC6 O25:H4 R SRRSR
VB8 rectal swab/C Sp M15 1 B2 EC6 O25:H4 R RRSSS
VB9 rectal swab/C Sp M15 1 B2 EC6 O25:H4 R RSRSR
HBS1 urine/H Sp M15 1 B2 EC6 O25:H4 R RRRSR
HBS4 ascites/H Sp M15 1 B2 EC6 O25:H4 R SSSSS
TNN
(TE2)
urine/H Ep-AC-1 M15 1 B2 EC6 O25:H4 R RSRSS
TE1 urine/H Ep-LTC-1 M15 1 B2 EC6 O25:H4 RR R R S S
HDE1 urine/H Ep-LTC 2a M15 1 B2 EC6 O25:H4 R SSSSS
HDE2 urine/H Ep-LTC-2b M15 1 B2 EC6 O25:H4 R RRSSR
HDE3 urine/H Ep-LTC-2c M15 1 B2 EC6 O25:H4 R SRSSR
Switzerland
EcS1 urine/NA Sp M15 5 B2 EC30 NT S S S S S S
3756
EcS2
urine/NA Sp M15 1 B2 EC6 O25:H4 R R R R S S
Spain
FV7561 urine, blood/H Sp M15 1 B2 EC6 O25:H4 R S R R S R
FV7563 urine/C Sp M15 1 B2 EC6 O25:H4 R S R R S R
FV7569,
FV7588,
FV7593,
FV7595
urine/H
Sp M15 1 B2 EC6 O25:H4 R S R R S R
FV7591 urine/H Sp M1 2 D EC68 O25:H4 S R S R S S
Portugal
5753 urine/C Sp M15 1 B2 EC6 O25:H4 R S R R S S
5754 blood/C Sp M15 1 B2 EC6 O25:H4 R R R R S S
5800 sputum/H Sp M15 1 B2 EC6 O25:H4 R R R R S S
5936 urine/C Sp M15 1 B2 EC6 O25:H4 R R R R S S
6373 urine/C Sp M15 1 B2 EC6 O25:H4 R R R R S R
Korea
KUMC
KN1604
urine/H Sp M15 1 B2 EC6 O25:H4 R R R R R R
KUMC
KN1608
urine/H Sp M15 6 A EC68 NT R R R R R R
India
E. coli 1 urine/H Sp M15 3 A EC66 O101 R S R R S S
E. coli 2 urine/H Sp M15 4 B1 EC74 NT R R R R R R
Lebanon
AH8,
AH9,
AH10,
AH15
faeces/C Sp M15 1 B2 EC6 O25:H4 R S R R S S
Canada
1100 urine/C Sp M15 1 B2 EC6 O25:H4 R RRRSS
17102 urine/C Sp M15 1 B2 EC6 O25:H4 R SRRSR
Continued
Disseminated E. coli clone ST131
275
at Universidad de Santiago de Compostela on February 6, 2016http://jac.oxfordjournals.org/Downloaded from
Sequence type (ST) determination
Multilocus sequence typing (MLST) was carried out as pre-
viously described.
26
Gene amplification and sequencing were
performed by using the primers specified at th e E. coli MLST
web site (http://web.mpiib-berlin.mpg.de/mlst/dbs/Ecoli) except
for mdh, icd and recA. Both the forward and reverse strands of
mdh were sequenced using primers mdh SF:
5
0
-CCAGGCGCTTGCACTACTGTTAA-3
0
and mdh SR:
5
0
-GCGATATCTTTCTTCAGCGTATC-3
0
, respectively, whereas
the forward strand of icd and the reverse strand of recA were
sequenced with the primers icd SF: 5
0
-CGGCAAACTCAAC
GTTCC-3
0
and recA SR: 5
0
-CTGACGCTGCAGGTGAT-3
0
,
respectively. Allelic profile and ST determinations were as per
the E. coli MLST web site scheme.
PFGE profiles
XbaI PFGE analysis was performed as previously described.
27
Profiles were compared digitally using BioNumerics software
(Applied Maths). Cluster analysis of Dice similarity indices
based on the unweighted pair group method with arithmetic
mean (UPGMA) was used to generate a dendrogram describing
the relationships among PFGE profiles. Isolates were considered
to belong to the same PFGE group if their Dice similarity index
was 85%.
28
Virulence genotypes
Forty-six extraintestinal virulence-associated genes were
detected by multiplex PCR, as previously described.
29
These
included 16 adhesin-encoding genes ( papAH, papC, papEF,
papG and its 3 alleles, sfa/focDE, sfaS, focG, afa/draBC, afaE8,
iha, bmaE, gafD, F17, clpG, fimH and hra), 8 toxin-encoding
genes (hlyA, hlyF, cnf1, cdtB, sat, pic, tsh and astA) and 4
siderophore-related genes (iroN, fyuA, ireA and iutA). They also
included 10 protectin/invasin-encoding genes (kpsM II, kpsMT
III, the K1, K2, K5 and K15 kps variants, rfc, traT, ibeA and
iss) and 7 pathogenicity island markers and miscellaneous genes
(cvaC, usp, ompT, clbB, clbN, fliC H7 and malX). A
UPGMA-based dendrogram was constructed depicting similarity
relationships among the isolates according to composite viru-
lence gene profiles.
Antibiotic susceptibility
Susceptibility to the following non-b-lactam molecules was
determined by disc diffusion: ciprofloxacin, gentamicin, amika-
cin, tetracycline, chloramphenicol and co-trimoxazole. Isolates
were defined as resistant or susceptible according to the stan-
dards of the French Antibiogram Committee.
30
Results
The primary strain set comprised 13 epidemiologically diverse
French E. coli isolates, all from the Paris area except one (strain
MECB5, from the south of France). All were known to be
characterized in terms of CTX-M-15 production, phylogenetic
group B2, ERIC2 PCR profile 1, serogroup O25, AmpC variant
6 and ciprofloxacin resistance (Table 1). H antigen determined
in this study was found to be H4.
These characteristics (when unknown) were newly assessed
for 28 other ESBL-positive E. coli isolates from seven other
countries, representing three continents. Twenty-three (82%) of
these 28 isolates were found to be identical to the 13 French iso-
lates with respect to all 6 core characteristics (CTX-M-15, group
B2, ERIC2 PCR profile 1, serotype O25:H4, AmpC EC6 and
ciprofloxacin resistance). These 23 isolates included 6 (86%) of
7 from Spain and 17 (81%) of 21 from the other six countries,
including all 15 from Lebanon, Portugal and Canada, plus 1
each from Switzerland and Korea (Table 1).
In contrast, five of the isolates (both isolates from India, and
one each from Spain, Switzerland and Korea) were found to
exhibit non-1 ERIC profiles (Figure 1) and non-EC6 AmpC
types, and proved to be mostly non-O25 and non-B2 (Table 1).
Moreover, the Spanish isolate (FV7591) exhibited CTX-M-1
rather than CTX-M-15 (Table 1).
These findings suggested that most (82%) of the non-French
isolates, like the 13 French isolates, represented a geographically
dispersed, group B2-derived, serotype O25:H4, AmpC variant
EC 6 clonal group of E. coli, characterized by CTX-M-15 and
ciprofloxacin resistance.
MLST results
To more rigorously assess phylogenetic relationships within
this collection, all 37 O25:H4 isolates (including the 36
Table 1. Continued
Country/
isolate
Sample/
acquisition site
Epidemic
(Ep) or
sporadic
(Sp) CTX-M-type
ERIC2
PCR
profile
Phylogenetic
group
AmpC
type Serotype
Antimicrobial susceptibility
CIP GEN AMK TET CHL SXT
15802 intra abd pus/H Sp M15 1 B2 EC6 O25:H4 R SSRSR
19502 urine/NH Sp M15 1 B2 EC6 O25:H4 R RSRSR
8501 urine/NH Sp M15 1 B2 EC6 O25:H4 R RRRSS
16102 urine/C Sp M15 1 B2 EC6 O25:H4 R SSSSS
R, resistant; S, susceptible; C, community-acquired; H, hospital-acquired; NH, nursing home; intra abd, intra-abdominal; AC, acute care; LTC, long-term care;
NA, not available; NT, non-typeable; CIP, ciprofloxacin; GEN, gentamicin; AMK, amikacin; TET, tetracycline; CHL, chloramphenicol; SXT, co-trimoxazole.
Italic font indicates the bacteriological characteristics that were already known when the strains were selected for this study.
Nicolas-Chanoine et al.
276
at Universidad de Santiago de Compostela on February 6, 2016http://jac.oxfordjournals.org/Downloaded from
CTX-M-15-positive isolates and the single CTX-M-1 isolate
from Spain) underwent seven-locus MLST. Irrespective of geo-
graphical origin, the 36 CTX-M-15-positive O25:H4 isolates
exhibited the same combination of alleles across the seven
sequenced loci, corresponding to an established ST, ST131. In
contrast, the Spanish CTX-M-1-positive O25:H4 isolate ( phylo-
genetic group D, ERIC2 PCR profile 2, ciprofloxacin-
susceptible) exhibited a novel combination of alleles and was
assigned to a new ST, ST648. This confirmed the clonality and
distinctness of the CTX-M-15 isolates.
PFGE profiles
Finer resolution of clonal relationships was obtained by PFGE
analysis. Figure 2 shows PFGE analysis results for the 36 ST131
strains and the ST648 Spanish strain FV7591. The 36 ST131
strains constituted one large cluster (defined at the 68% simi-
larity level), which was tied to the ‘outgroup’ strain FV7591 at
,40% similarity. The ST131 cluster, in turn, comprised 17 sep-
arate PFGE groups, as defined at the 85% similarity level. These
PFGE groups corresponded inconsistently with geographical
origin. That is, the 13 French strains were classified into seven
PFGE groups, the 6 Canadian strains into five groups, the 5
Portuguese strains into four groups and the 4 Lebanese strains
into two groups. The six Spanish CTX-M-15-producing strains
represented the only example of all isolates from a given
country being classified into the same PFGE group. (Of note,
the single Korean strain and the single Swiss strain were the sole
representatives of their respective PFGE groups.) Likewise,
multiple countries were represented within cer tain PFGE groups,
including PFGE group I (France and Canada), group V (France,
Canada and Portugal) and group XIII (France and Portugal).
Nonetheless, frankly indistinguishable PFGE profiles were
encountered only among strains from the same country, includ-
ing two strains each from France (VB6 and HBS1), Lebanon
(AH15 and AH10) and Spain (FV7569 and FV7595).
Virulence profiles
Extended virulence profiles were determined for the 36 ST131
isolates to assess the extent of within-group diversity and the
virulence potential of the clonal group. Of the 46 virulence
genes tested, 16 (35%) were detected in at least 1 isolate each.
Isolates contained from 7 to 14 genes each (Table 2). Five
different virulence genes were uniformly present in all 36
isolates, including fimH (type I fimbriae), sat (secreted auto-
transporter toxin), fyuA (yersiniabactin receptor), usp (uropatho-
genic specific protein) and malX (pathogenicity island marker)
(Table 2). Four other genes were present in .90% of the iso-
lates, including iha (adhesin-siderophore receptor: 91%), kpsM
II (group 2 capsule synthesis: 94%), iutA (aerobactin receptor:
97%) and ompT (outer membrane protease T: 97%) (Table 2),
with the K5 and K2 kpsM II variants being detected in 53% and
39% of the isolates, respectively. Intermediate prevalence viru-
lence genes included traT (serum resistance associated: 75%)
and afa/draBC (afimbrial Dr-binding adhesins: 22%). In con-
trast, three genes occurred in ,12% of strains each , typically
together. These included hlyF (haemolysin F: 8%), iss
(increased serum survival: 8%) and iroN (siderophore receptor:
11%) (Table 2).
Overall, virulence profile similarity among the 36 isolates
was high, ranging from 91% to 100% (Figure 3). Only eight iso-
lates exhibited a unique virulence profile. Indeed, 14 isolates
(from Canada, France, Portugal, Korea and Switzerland) had an
identical 11-gene virulence profile and four other groups of 2–6
strains each exhibited uniform VF profiles (Figure 3 and
Table 2). Although some geographical segregation of virulence
profiles was evident, virulence profiles corresponded inconsist-
ently with PFGE type or locale (Figure 3), suggesting ongoing
evolution of virulence genotypes.
Antimicrobial susceptibility patterns
To assess the multidrug resistance of the 36 ST131 isolates, sus-
ceptibility to non-b-lactam antimicrobials was tested (Table 1).
Eighty-three per cent of the isolates were resistant to tetra-
cycline, 77% to amikacin, 53% to co-trimoxazole and 50% to
gentamicin, but only 0.3% to chloramphenicol.
Discussion
Our findings provide novel evidence of a recently emerged,
broadly dissemin ated, CTX-M-15-positive E. coli clonal group
as a cause of mult idrug-resistant extraintestinal infections on at
least three continents. This lineage exhibits a fair ly robust viru-
lence gene profile, implying substantial extraintestinal patho-
genic potential. In most study locales, it accounted for a large
proportion of ESBL-p ositive E. coli that were CTX-M-15 and/or
O25-positive. The emergence of a new multidrug-resistant
Figure 1. ERIC2 PCR profiles of seven Spanish and three French isolates
of ESBL-producing E. coli. Lane 1, molecular weight marker. Lanes 2 –8,
Spanish isolates with strain FV7591 in lane 6. Lanes 9– 11, French isolates
[strain HBS1 (lane 9), strain HDE2 (lane 10) and strain TE1 (lane 11)].
A uniform profile was found among these isolates except for the Spanish
strain FV7591 (CTX-M-1).
Disseminated E. coli clone ST131
277
at Universidad de Santiago de Compostela on February 6, 2016http://jac.oxfordjournals.org/Downloaded from
extraintestinal pathogen that may be spreading rapidly through
the population while continuing to evolve appears to pose a sig-
nificant public health threat in need of urgent attention.
The clonality of the ST131 strains is evident from their hom-
ogeneity with respect to phylogenetic group, seven-gene MLST
allele combination and ERIC2 PCR profile. Clonality is further
supported by the isolates’ uniform serotype (O25:H4),
b-lactamase repertoire (CTX-M-15 and AmpC EC6) and cipro-
floxacin phenotype, and their .90% similar virulence gene pro-
files. Furthermore, the considerable similarity of PFGE profiles
observed among certain isolates indicates quite recent diver-
gence from a common ancestor, whereas the occurrence in
different locales of isolates with similar PFGE profiles suggests
recent or ongoing transmission.
Figure 2. XbaI-PFGE dendrogram for 36 CTX-M-15-positive E. coli isolates from ST131 and a Spanish strain from ST648. The dendrogram for the 37
isolates, as produced by the UPGMA algorithm based on Dice similarity coefficients, included 18 PFGE groups, as defined based on 85% similarity of
PFGE profiles.
Nicolas-Chanoine et al.
278
at Universidad de Santiago de Compostela on February 6, 2016http://jac.oxfordjournals.org/Downloaded from
The virulence of the O25:H4-ST131 isolates can be inferred
from two lines of evidence. First, most isolates were from
samples submitted to clinical microbiology laboratories from
inpatients and outpatients, so likely caused extraintestinal infec-
tions; this certainly was true for the blood and ascites isolates.
Second, the number and types of virulence genes present in
these strains (7–14 per isolate; coding for adhesins, sidero-
phores, toxins, protectins and pathogenicity island markers)
imply a robust virulence capability.
29,31
Although these viru-
lence profiles are not so extensive as those of typical
antimicrobial-susceptible pathogenic E. coli from phylogenetic
group B2, they nonetheless are more extensive than is usually
observed among fluoroquinolone- or extended-spectrum
cephalosporin-resistant E. coli, including human clinical iso-
lates.
32
Thus, these strains appear to pose the double threat of
multidrug resistance (including to first-line therapeutic agents
for Gram-negative infections) and substantial extraintestinal
virulence capability. This makes their emergence and
dissemination particularly concerning and sugg ests a need to
identify their origins, reservoirs and transmission pathways so
that appropriate interventions can be implemented. Better defi-
nition of the extent of this problem is needed, to clarify how
great a public health threat these strains actually pose, so that
resources can be allocated accordingly.
Dissemination of antimicrobial resistance genes by spread of
the particular clone(s) in which they reside differs from the
established paradigm for the emergence of ESBLs, which
involves transfer of resistance-encoding plasmids rather than the
host bacteria per se.
9
However, clonal dispersal of drug-resistant
pathogens has precedent in other species, such as methicillin-
resistant S. aureus and penicillin-resistant S. pneumoniae.
11,12
It
also has been documented in E. coli, as exemplified by the loca-
lized outbreaks and international dissemination observed with
multidrug-resistant clonal groups such as (group D-derived)
‘clonal group A’ and serotype O15:K52:H1, including the recent
detection of clonal group A isolates in wastewater effluents from
Table 2. Virulence genotype of 36 CTX-M-15-producing Escherichia coli strains of clone ST131
Country/isolate
Adhesin Toxin Siderophore
Protectin, invasin, pathogenicity island marker,
miscellaneous
afa/
draBC iha fimH hlyF sat iroN fyuA iutA
kpsM
II
K5 kps
variant
K2 kps
variant usp traT ompT iss malX
France
MECB5 2 þþ þþþ þþ þ þ 2 þþ þ þ þ
VA1 2 þþ þþþ þþ þ þ 2 þþ þ 2 þ
Vlab2, VB8, HBS4, TNN
(TE2), TE1, HDE1
2 þþ 2 þ 2 þþ þ þ 2 þþ þ 2 þ
VB9 2 þþ 2 þ 2 þþ þ þ 2 þ 222þ
HDE2 2 þþ 2 þ 2 þþ þ 22þþ þ 2 þ
HDE3 22þ 2 þ 2 þþ þ 2 þþþþ2 þ
VB6, HBS1 þþþ2 þ 2 þþ þ 2 þþ2 þ 2 þ
Switzerland
3756 EcS2 2 þþ 2 þ 2 þþ þ þ 2 þþ þ 2 þ
Spain
FV7561, FV7563 þþþ2 þ 2 þþ þ 2 þþþþ2 þ
FV7569, FV7588,
FV7593, FV7595
þþþ2 þ 2 þþ þ 2 þþ2 þ 2 þ
Portugal
5753 22þ 2 þ 2 þ 22 2 2 þþ þ 2 þ
5936 22þ 2 þ 2 þþ 22 2þþ þ 2 þ
5754, 5800, 6373 2 þþ 2 þ 2 þþ þ þ 2 þþ þ 2 þ
Korea
KUMC KN1604 2 þþ 2 þ 2 þþ þ þ 2 þþ þ 2 þ
Lebanon
AH9, AH8, AH15, AH10 2 þþ 2 þ 2 þþ þ 2 þþþþ2 þ
Canada
1100, 8501, 16102 2 þþ 2 þ 2 þþ þ þ 2 þþ þ 2 þ
17102 2 þþ 2 þ 2 þþ þ 2 þþ
2 þ 2 þ
15802 2 þþ 2 þþ
þ þ þ þ 2 þ 2 þþþ
19502 2 þþ þþþ þ þ þ þ 2 þþ þ þ þ
afa/draBC, afimbrial Dr-binding adhesins; iha, adhesin-siderophore receptor; fimH, type I fimbriae; hlyF, haemolysin F; sat, secreted auto-transporter toxin;
iroN, siderophore receptor; fyuA, yersiniabactin receptor; iutA, aerobactin receptor; kpsM II, group 2 capsule synthesis (variant K5 and K2); usp,
uropathogenic specific protein; traT, serum resistance associated; ompT, outer membrane protease; iss, increased serum survival; malX, pathogenicity island
marker.
Disseminated E. coli clone ST131
279
at Universidad de Santiago de Compostela on February 6, 2016http://jac.oxfordjournals.org/Downloaded from
geographically dispersed areas of the United States.
10,13,33
The
present CTX-M-15-positive E. coli clonal group was previously
shown to have caused what appeared to be localized outbreaks
involving specific healthcare institutions (France) or geographi-
cal regions (Calgary).
20,21
Our findings suggest that some of
these seemingly isolated occurrences are actually linked, a prin-
ciple that may apply broadly to drug-resistant extraintestinal
infections. From this point of view, it would be relevant to
determine whether the serogroup O25, CTX-M-15-positive
E. coli previously published, notably in the UK, and not
included in this study also belong to clone ST131.
34
Recognition
that geographically distant infection episodes may be caused by
the same bacterial clone, arising from a common source, is the
basis for the CDC’s PulseNet surveillance system.
35
Whereas
that system focuses mainly on diarrhoeal pathogens, a similar
system may be needed for extraintestinal infections.
In summary, we have characterized a broadly disseminated,
CTX-M-15-positive, multidrug-resistant, virulent E. coli clonal
group with highly homogeneous virulence genotypes and sub-
groups exhibiting highly similar PFGE profiles, suggesting
recent emergence. Understanding how this clone has emerged
and successfully disseminated within the hospital and commu-
nity, including across national boundaries, should be a public
health priority.
Acknowledgements
We are indebted to Professor Patrice Nordmann, Professor
Guillaume Arlet and Dr Florence Doucet-Populaire for providing
us with the Indian and Swiss strains, strain TNN (TE2) and
the Lebanese strains, respectively. We also are grateful to
Dr Azucena Mora, Dr Jesus Blanco, Dr Miguel Blanco,
Mrs Ghizlane Dahbi and Mrs Cecilia Lopez for their contri-
bution to this study.
Funding
This study was supported by a grant (AOR 04016) from La
Direction de le Recherche Clinique de l’Assistance
Publique-Ho
ˆ
pitaux de Paris (M.-H. N.-C.), a grant
(PI052023-PI051481) from Fondo de Investigacion Sanitaria
(FIS), Instituto de Salud Carlos III, Spanish Ministerio de
Sanidad y Consumo (J. B.) and Office of Research an d
Development, Medical Research Service, Department of
Veterans Affairs (J. R. J.).
Figure 3. Virulence profile dendrogram for 36 CTX-M-15-positive E. coli isolates from ST131. The dendrogram was produced by the UPGMA algorithm
based on extended virulence gene profiles for the 36 strains from ST131. Virulence profile similarity varied from 91% to 100%.
Nicolas-Chanoine et al.
280
at Universidad de Santiago de Compostela on February 6, 2016http://jac.oxfordjournals.org/Downloaded from
Transparency declarations
All of the authors except one have none to declare. J. R. J. is a
consultant for the following companies: Bayer, Ortho-McNeil,
Merck, Wyeth-Ayerst, and Procter and Gamble.
References
1. Paterson D, Bonomo R. Extended-spectrum b-lactamases: a
clinical update. Clin Microbiol Rev 2005; 18: 657–86.
2. Pitout JD, Nordmann P, Laupland KB et al. Emergence of
Enterobacteriaceae producing extended-spectrum b-lactamases
(ESBLs) in the community. J Antimicrob Chemother 2005; 56:52–9.
3. Canton R, Coque TM. The CTX-M b-lactamase pandemic. Curr
Opin Microbiol 2006; 9: 466 –75.
4. Bonnet R. Growing group of extended-spectrum b-lactamases:
the CTX-M enzymes. Antimicrob Agents Chemother 2004; 48: 1 –14.
5. Gangoue-Pieboji J, Miriagou V, Vourli S et al. Emergence of
CTX-M-15-producing enterobacteria in Cameroon and characterization
of a bla
CTX-M-15
carrying element. Antimicrob Agents Chemother 2005;
49: 441–5.
6. Lavollay M, Mamlouk K, Frank T et al. Clonal dissemination of a
CTX-M-15 b-lactamase-producing Escherichia coli strain in the Paris
area, Tunis and Bangui. Antimicrob Agents Chemother 2006; 50:
2433–8.
7. Livermore DM, Canton R, Gniadkowski M et al. CTX-M: chan-
ging the face of ESBLs in Europe. J Antimicrob Chemother 2007; 59:
165–74.
8. Pallecchi L, Malossi M, Mantella A et al. Detection of
CTX-M-type b-lactamase genes in fecal Escherichia coli isolates from
healthy children in Bolivia and Peru. Antimicrob Agents Chemother
2004; 48: 4556 –61.
9. Fierer J, Guiney D. Extended-spectrum b-lactamases: a plague
of plasmids. JAMA 1999; 281: 563 –4.
10. Manges AR, Johnson JR, Foxman B et al. Widespread distri-
bution of urinary tract infections caused by a multidrug-resistant
Escherichia coli clonal group. N Engl J Med 2001; 345: 1007–13.
11. McGee L, McDougal L, Zhou J et al. Nomenclature of major
antimicrobial-resistant clones of Streptococcus pneumoniae defined by
the pneumococcal molecular epidemiology network. J Clin Microbiol
2001; 39: 2565 –71.
12. Oliveira DC, Alexander T, de Lencastre H. Secrets of success of a
human pathogen: molecular evolution of pandemic clones of methicillin-
resistant Staphylococcus aureus. Lancet Infect Dis 2002; 2:180–9.
13. Prats G, Navarro F, Mirelis B et al. Escherichia coli serotype
O15:K52:H1 as a uropathogenic clone. J Clin Microbiol 2000; 38:
201–9.
14. Arpin C, Coulange L, Dubois V et al. Extended-spectrum
b
-lactamase-producing Enter
obacteriaceae in various types of private
health care centers. Antimicrob Agents Chemother 2007; 51: 3040–4.
15. Prince N, Leflon-Guibout V, Doit C et al. Genetic background of
CTX-M-producing Escherichia coli isolates reveals a great strain diver-
sity and the emergence of a clone in the community and the hospital.
In: Abstracts of the Forty-sixth Interscience Conference on
Antimicriobial Agents Chemotherapy, San Francisco, CA, 2006.
Abstract K-1508, p. 357. American Society for Microbiology,
Washington, DC, USA.
16. Karim A, Poirel L, Nagarajan S et al. Plasmid-mediated
extended-spectrum b-lactamase (CTX-M-3 like) from India and gene
association with insertion sequence ISEcp1. FEMS Microbiol Lett
2001; 201: 237 –41.
17. Kim Y, Kim S, Lee J et al. Nosocomial transmission of
CTX-M-15 and OXA-30 b-lactamase-producing Escherichia coli in a
neurosurgical intensive care unit. Ann Clin Lab Sci 2005; 35: 297 –301.
18. Mendonc¸a N, Leitao J, Manageiro V et al. Spread of clinical
extended-spectrum b-lactamase (CTX-M)-producing Escherichia coli
isolates in community and nosocomial environments in Portugal.
Antimicrob Agents Chemother 2007; 51: 1946–55.
19. Moubareck C, Daoud Z, Hakime
´
NI et al. Countrywide spread of
community- and hospital-acquired extended-spectrum b-lactamase
(CTX-M-15)-producing Enterobactericeae in Lebanon. J Clin Microbiol
2005; 43: 3309 –13.
20. Pitout JDD, Laupland KB, Church DL et al. Virulence factors of
Escherichia coli isolates that produce CTX-M-type extended-spectrum
b-lactamases. Antimicrob Agents Chemother 2005; 49: 4667–70.
21. Leflon-Guibout V, Jurand C, Bonacorsi S et al. Emergence and
spread of three clonally related virulent isolates of CTX-M-15-producing
Escherichia coli with variable resistance to aminoglycosides and
tetracycline in a French geriatric hospital. Antimicrob Agents Chemother
2004; 48: 3736–42.
22.
Johnson JR, O’Bryan TT. Improved repetitive-element PCR fin-
gerprinting
for resolving pathogenic and nonpathogenic phylogenetic
groups within Escherichia coli. Clin Diagn Lab Immunol 2000; 7:
265–73.
23. Clermont O, Bonacorsi S, Bingen E. Rapid and simple determi-
nation of the Escherichia coli phylogenetic group. Appl Environ
Microbiol 2000; 66: 4555– 8.
24. Guine
´
e PAM, Jansen WH, Wadstro
¨
mTet al. Escherichia coli
associated with neonatal diarrhoea in piglets and calves. In: Leeww
PW, Guine
´
e PAM, eds. Laboratory Diagnosis in Neonatal Calf and
Pig Diarrhoea: Current Topics in Veterinary and Animal Science. The
Hague, Netherlands: Martinus-Nijhoff, 1981; 126– 62.
25. Clermont O, Johnson JR, Menard M et al. Determination of
Escherichia coli O types by allele-specific polymerase chain reaction:
application to the O types involved in human septicemia. Diagn
Microbiol Infect Dis 2007; 57: 214 –23.
26. Tartof SY, Solberg OD, Manges AR et al. Analysis of a uro-
pathogenic Escherichia coli clonal group by multilocus sequence
typing. J Clin Microbiol 2005; 43: 5860–4.
27. Blanco M, Blanco JE, Dahbi G et al. Typing of intimin (eae)
genes from enteropathogenic Escherichia coli (EPEC) isolated from
children with diarrhoea in Montevideo, Uruguay: identification of two
novel intimin variants (mB and jR/b2B). J Med Microbiol 2006; 55:
1165–74.
28. Carrico JA, Pinto FR, Simas C et al. Assessment of band-based
similarity coefficients for automatic type and subtype classification of
microbial isolates analyzed by pulsed-field gel electrophoresis. J Clin
Microbiol 2005; 43: 5483– 90.
29. Johnson JR, Kuskowski MA, O’Bryan TT et al. Epidemiological
correlates of virulence genotype and phylogenetic background among
Escherichia coli blood isolates from adults with diverse-source bactere-
mia. J Infect Dis
2002; 185:
1439–47.
30. Anonymous. Comite
´
de l’Antibiogramme de la Socie
´
te
´
Franc¸aise
de Microbiologie report 2003. Int J Antimicrob Agents 2003; 21:
364–91.
31. Johnson JR, Clermont O, Menard M et al. Experimental mouse
lethality of Escherichia coli isolates in relation to accessory traits, phy-
logenetic group, and ecological source. J Infect Dis 2006; 194:
1141–50.
32. Johnson JR, Kuskowski MA, Owens K et al. Phylogenetic origin
and virulence genotype in relation to resistance to fluoroquinolones
and/or extended-spectrum cephalosporins and cephamycins among
Escherichia coli isolates from animals and humans. J Infect Dis 2003;
188: 759– 68.
Disseminated E. coli clone ST131
281
at Universidad de Santiago de Compostela on February 6, 2016http://jac.oxfordjournals.org/Downloaded from