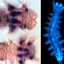
Hox genes are transcriptional regulators that elicit cell positional identity along the anterior-posterior region of the body plan across different lineages of Metazoan. Comparison of Hox gene expression across distinct species reveals their evolutionary conservation, however their gains and losses in different lineages can correlate with body plan modifications and morphological novelty. We compare the expression of eleven Hox genes found within Streblospio benedicti , a marine annelid that produces two types of offspring with distinct developmental and morphological features. For these two distinct larval types, we compare Hox gene expression through ontogeny using HCR (hybridization chain reaction) probes for in-situ hybridization and RNA-seq data. We find that Hox gene expression patterning for both types is typically similar at equivalent developmental stages. However, some Hox genes have spatial or temporal differences between the larval types that are associated with morphological and life-history differences. This is the first comparison of developmental divergence in Hox genes expression within a single species and these changes reveal how body plan differences may arise in larval evolution.